Définitions
L’AMP vigilance a pour objet la surveillance des incidents relatifs aux gamètes, aux tissus germinaux et aux embryons utilisés à des fins d’assistance médicale à la procréation ou à des fins de préservation de la fertilité, ainsi que des effets indésirables observés chez les donneurs de gamètes ou chez les personnes qui ont recours à l’assistance médicale à la procréation.
On entend par incident tout accident ou erreur survenant au cours des différentes étapes du processus d’AMP, susceptible d’entraîner un effet indésirable sur la personne ou la perte de gamètes, de tissus germinaux ou d’embryons. À titre d’exemple, des incidents à type de perte partielle ou totale d’embryons liée à un matériel défectueux (ex : panne de congélateur) ou de problèmes d’étiquetage des tubes de liquide folliculaire ont été rapportés.
On entend par effet indésirable toute réaction nocive survenant chez une personne, liée ou susceptible d’être liée aux activités d’AMP au cours de ses différentes étapes (stimulation, ponction folliculaire, insémination, transfert embryonnaire). À titre d’exemple, des effets indésirables à type d’hyperstimulations ovariennes sévères, d’hémopéritoine, et d’accidents thromboemboliques ont été rapportés.
Dispositif d’AMP vigilance
L’AMP vigilance est une vigilance sanitaire réglementée confiée à l’Agence de la biomédecine par la loi n°2004-800 du 6 août 2004 relative à la bioéthique. Le décret de juin 20081 a complété l’organisation du dispositif et précisé les rôles des différents acteurs. La loi n°2011-814 du 7 juillet 2011 relative à la bioéthique n’a pas introduit de modifications dans ce champ.
Chaque centre autorisé pour les activités d’AMP doit désigner un correspondant local d’AMP vigilance (CLA). Ce dernier a pour missions de recueillir tous les incidents et les effets indésirables, de les déclarer sans délai à l’Agence de la biomédecine, d’informer les autres correspondants locaux d’AMP vigilance si nécessaire et les autres vigilances sanitaires concernées de son établissement, de participer aux investigations, d’aviser l’Agence de la biomédecine des résultats des investigations et en cas de difficultés de fonctionnement du dispositif. Tout autre professionnel qui ne dépend pas d’un centre d’AMP et qui a connaissance de la survenue d’un incident et d’un effet indésirable doit en informer sans délai l’Agence de la biomédecine. En pratique, il est préférable que ce professionnel de santé en informe le centre d’AMP dans lequel le couple a été pris en charge initialement et que le CLA fasse la déclaration de l’incident et de l’effet indésirable.
Fin 2013, le réseau d’AMP vigilance comprend 191 correspondants locaux d’AMP vigilance désignés dans 96% des 199 centres d’AMP (103 centres clinico-biologiques et 96 laboratoires pratiquant uniquement l’insémination artificielle2).
Les déclarations d’AMP vigilance sont évaluées et expertisées par l’Agence de la biomédecine en lien avec un groupe de travail externe “AMP vigilance” composé d’experts biologistes et gynécologues-obstétriciens spécialisés dans le domaine de l’AMP. L’Agence de la biomédecine a la possibilité de proposer ou de mettre en œuvre des mesures préventives et/ou correctives dans un but de réduction des risques et d’amélioration des pratiques. L’Agence de la biomédecine assure le secrétariat de la commission nationale d’AMP vigilance.
L’AMP vigilance comporte une dimension transversale forte, impliquant souvent d’autres systèmes de vigilance sanitaire, notamment la matériovigilance et la pharmacovigilance. Ceci a justifié le développement de procédures d’échanges des données avec ces systèmes de vigilance sanitaire pilotés par l’ANSM.
Matériel et méthodes
Les signalements sont transmis à l’Agence de la biomédecine par les CLA au moyen de la fiche de déclaration, de préférence par l’application informatique sécurisée AMP Vigie.
En 2013, 92% des déclarations ont été saisies directement en ligne par le CLA du centre d’AMP.
Les incidents et effets indésirables recueillis sont analysés et évalués en termes de gravité et d’imputabilité :
On distingue ainsi 5 niveaux de gravité, de G1 à G5, présentés ci-dessous :
| G1 | Diminution de la performance du processus, sans conséquence sur son résultat, et/ou source de contrainte opérationnelle acceptable |
| G2 | Dégradation de la performance du processus susceptible ou ayant altéré de façon modérée son résultat et/ou source de contrainte opérationnelle non acceptable Perte d’embryons et/ou de gamètes sans disparition des chances de procréation sur la tentative |
| G3 | Dégradation de la performance du processus ayant altéré de façon importante son résultat. Complications liées au processus d’AMP avec hospitalisation et/ou incapacité fonctionnelle mineure Intervention médicale ou chirurgicale afin d’exclure tout dommage permanent ou infirmité corporelle Risque de transmission d’affection(s) à morbidité modérée accessible(s) à un traitement Perte d’embryons et/ou des gamètes avec disparition des chances de procréation sur la tentative |
| G4 | Acte ou procédure sur un patient autre (erreur d’attribution) Perte d’embryons et/ou des gamètes avec disparition définitive des chances de procréation pour le couple Complications sévères liées au processus d’AMP avec hospitalisation supérieure à 7 jours et/ou incapacité fonctionnelle majeure Risque de transmission par les gamètes d’affection(s) à morbidité sévère : affections transmissibles avec mise en jeu du pronostic vital |
| G5 | Décès au cours du processus d’AMP Incapacité fonctionnelle majeure et permanente |
L’imputabilité est une estimation individuelle pour une déclaration donnée de la probabilité de la relation existante entre le processus d’AMP et la survenue d’un incident ou d’un effet indésirable. L’ensemble des étapes du processus d’AMP et leur environnement doivent être pris en compte. Il peut y avoir une différence entre le niveau d’imputabilité établi lors de la survenue de l’événement indésirable et celui retenu après investigation du cas. Il s’agit donc d’une estimation initiale qui est réévaluée et modifiée si besoin par le CLA dans la partie B de la fiche de déclaration. Après réception de chaque déclaration, l’Agence de la biomédecine réévalue son niveau d’imputabilité pour s’assurer d’une utilisation cohérente de l’échelle ou attribue une imputabilité à cet événement si cela n’a pas déjà été fait par le déclarant.
Résultats
Entre le 1er janvier et le 31 décembre 20133, l’Agence de la biomédecine a reçu 469 déclarations d’AMP vigilance provenant de 86 centres d’AMP comprenant 81 centres clinico-biologiques et 5 laboratoires d’insémination artificielle.
En complément du nombre de déclaration, le nombre d’événements indésirables constatés chaque année a été calculé depuis l’année 2009.
L’analyse du nombre de déclaration par année permet d’évaluer l’efficacité du dispositif mis en place alors que l’analyse du nombre d’événements constatés permet d’évaluer la volumétrie d’événements indésirables par année.
L’évolution du nombre de déclarations d'AMP vigilance, du nombre d’événements constatés et du nombre de centres déclarants de 2009 à 2013 est présentée à la figure AMPV1.
Figure AMPV1. Evolution du nombre de déclarations d’AMP vigilance, du nombre d’évènements constatés et du nombre de centres déclarants de 2009 à 2013
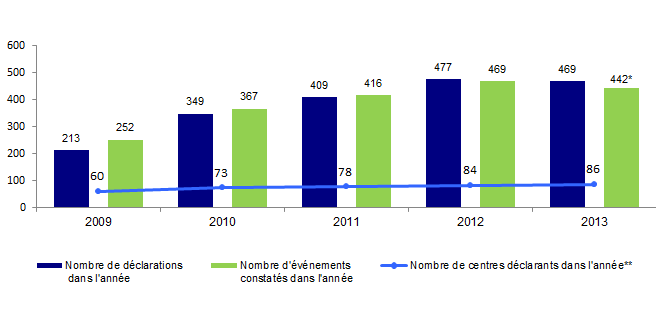
*Les déclarations reçues après le 14 février 2014 ne sont pas prises en compte (date du gel de la base). Entre le 1er janvier 2013 et le 14 février 2014, 442 incidents et effets indésirables constatés en 2013 ont été déclarés.
** Nombre de centres clinico-biologiques et laboratoires d'insémination artificielle
On observe que le nombre de déclarations a été multiplié par un facteur 2 entre 2009 et 2012. Par contre, le nombre de déclarations reste stable entre 2012 et 2013.
Parallèlement, le nombre de centres d’AMP clinico-biologiques déclarants a augmenté depuis 2009 passant de 54 % (58/107) à 79% en 2013 (81/103).
Pour les laboratoires d’insémination artificielle, le nombre de centres déclarants est beaucoup plus faible mais a également augmenté depuis 2009 passant de 2% (2/105) à 5% (5/96) en 2013.
Délais de déclaration
Le décret de juin 2008 prévoit que les CLA déclarent les incidents et les effets indésirables sans délai à l’Agence de la biomédecine.
La répartition du nombre cumulé d’événements constatés de 2009 à 2012 selon la date de déclaration est présentée à la figure AMPV2.
Figure AMPV2. Répartition du nombre cumulé d’évènements constatés de 2009 à 2013 selon la date de déclaration
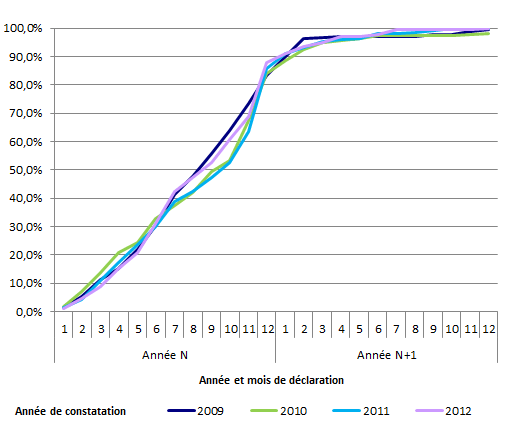
Entre 2009 et 2012, on observe que l’on a recueilli en données cumulées, à la fin de l’année civile, en moyenne 85 % des événements indésirables constatés dans l’année et que quasiment la totalité (98%) sont recueillis en milieu d’année n+1. Au cours des années, on observe une légère augmentation en fin d’année civile de l’exhaustivité des déclarations des événements constatés dans l’année (83 % en 2009 vs 88% en 2012).
L’année 2013 n’a pas été prise en compte par manque de recul en 2014 pour faire cette analyse.
L’évolution du délai de déclaration depuis 2009 et des délais de déclaration en fonction de la gravité sont présentés dans le tableau AMPV1.
Tableau AMPV1. Délais de déclaration des événements en jours en fonction de la gravité
Année de |
|
G1 à G5 |
G1 |
G2 |
G3 |
G4 |
G5 |
2009 |
. |
|
|
|
|
|
|
|
N* |
213 |
5 |
42 |
136 |
30 |
0 |
Délai (jours) |
Moyenne±Écart-type |
51±60 |
32±23 |
35±40 |
49±46 |
89±111 |
|
Médiane |
32 |
31 |
22 |
33 |
44 |
|
|
Extrêmes |
[0-501] |
[0-64] |
[1-203] |
[0-209] |
[9-501] |
|
|
2010 |
. |
|
|
|
|
|
|
|
N* |
349 |
21 |
54 |
203 |
71 |
0 |
Délai (jours) |
Moyenne±Écart-type |
77±83 |
91±117 |
72±92 |
76±78 |
78±79 |
|
Médiane |
46 |
24 |
34 |
48 |
55 |
|
|
Extrêmes |
[0-595] |
[0-434] |
[0-373] |
[0-595] |
[0-319] |
|
|
2011 |
. |
|
|
|
|
|
|
|
N* |
409 |
11 |
56 |
245 |
96 |
1 |
Délai (jours) |
Moyenne±Écart-type |
82±87 |
49±47 |
76±98 |
83±88 |
81±75 |
415±NC |
Médiane |
47 |
34 |
32 |
49 |
51 |
415 |
|
Extrêmes |
[0-487] |
[0-120] |
[0-363] |
[0-487] |
[0-314] |
NC |
|
2012 |
. |
|
|
|
|
|
|
|
N* |
477 |
27 |
48 |
282 |
119 |
1 |
Délai (jours) |
Moyenne±Écart-type |
86±107 |
35±53 |
49±45 |
94±115 |
90±107 |
468±NC |
Médiane |
48 |
12 |
34 |
50 |
55 |
468 |
|
Extrêmes |
[0-798] |
[0-241] |
[1-184] |
[0-601] |
[0-798] |
NC |
|
2013 |
. |
|
|
|
|
|
|
|
N* |
469 |
34 |
79 |
256 |
100 |
0 |
Délai (jours) |
Moyenne±Écart-type |
66±89 |
31±36 |
67±94 |
62±86 |
86±101 |
|
Médiane |
36 |
19 |
32 |
34 |
54 |
|
|
Extrêmes |
[0-917] |
[0-140] |
[0-531] |
[0-917] |
[0-681] |
|
NC : non calculable
N*: nombre de déclarations
Le délai moyen entre la déclaration faite à l’Agence de la biomédecine en 2013 et la date de constatation de l’événement indésirable a été de 66 jours soit d’environ 2 mois avec une médiane à 36 jours. Il existe une grande variabilité de ce délai attestée par la valeur de l’écart-type à 89 jours et les valeurs extrêmes comprises entre 0 jour et 917 jours soit un délai de 2 ans et demi pour déclarer un événement indésirable à l’Agence de la biomédecine après sa constatation. Aucun élément fourni ne permettait d’expliquer ce long délai de déclaration.
En comparaison aux 3 années précédentes, on observe une tendance à la diminution du délai médian de déclaration.
En moyenne, le délai de déclaration est plus long pour des événements plus graves et plus complexes à documenter. Une explication possible est que dans ces situations les centres ont déclaré l’événement indésirable après avoir finalisé l’investigation bien que cette démarche ne soit pas conforme avec les exigences réglementaires.
Distribution des déclarations d’AMP vigilance par région administrative
La distribution des déclarations par région est présentée en fonction de la gravité sous forme d’un histogramme à la figure AMPV3. Il s’agit de données brutes qui ne sont pas rapportées à l’activité.Figure AMPV3. Répartition des déclarations par région selon la gravité des événements indésirables (2013, n= 469)
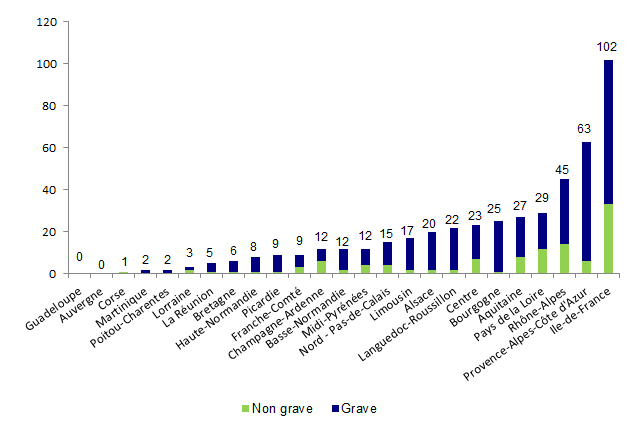
En 2013, 86 centres d’AMP (centres clinico-biologiques d’AMP ou laboratoires d’IA) répartis dans 23 régions ont fait au moins une déclaration d’AMP vigilance. Dans 2 régions (Guadeloupe et Auvergne), les centres d’AMP n’ont fait aucune déclaration d’AMP vigilance.
Dans les autres régions, en moyenne près de 20 déclarations d’AMP vigilance ont été faites par région avec des extrêmes allant de 1 déclaration à 102 déclarations.
On observe qu’au niveau de la région les déclarations concernent majoritairement des évènements indésirables graves.
Pour 12 régions, la totalité des centres clinico-biologiques de la région ont fait au moins une déclaration d’AMP vigilance en 2013.
La distribution du nombre de déclarations d’événements indésirables pour 1000 actes d’AMP par région est présentée à la figure AMPV4.
Figure AMPV4. Taux de déclarations d'événements indésirables (pour 1 000 actes d'AMP)
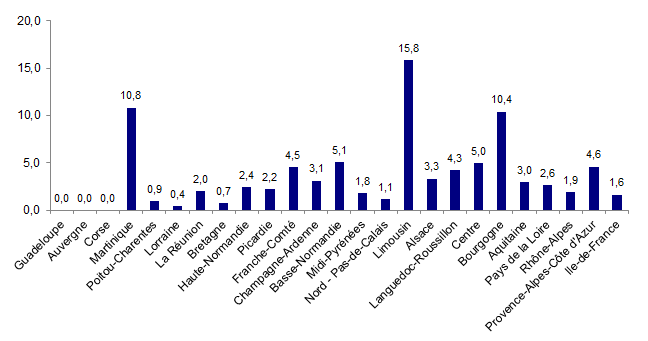
En rapportant le nombre de déclarations d’événements indésirables au nombre d’actes d’AMP4, la répartition des régions diffère. Les régions ayant une forte activité en AMP n’ont pas proportionnellement le plus fort taux de notification.
Distribution des incidents et des effets indésirables en fonction de la gravité
La gestion des déclarations par l’Agence de la biomédecine est notamment basée sur le niveau de gravité des conséquences des événements indésirables rapportés.
La distribution des incidents et des effets indésirables en fonction de la gravité est présentée dans la figure AMPV5.
Figure AMPV5. Nombre d’incidents et d’effets indésirables en fonction de la gravité (2013, n = 469)
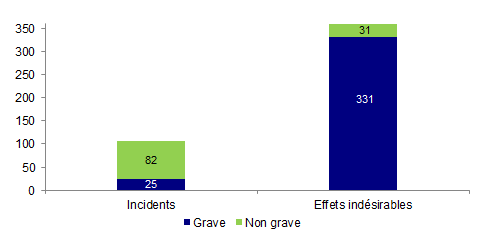
En 2013, 356 événements, soit près de 76 % des incidents et des effets indésirables, ont été cotés comme graves (G3, G4, G5).
On remarque que les effets indésirables sont pour la plupart (91 %) cotés comme graves alors que les incidents sont le plus souvent cotés comme non graves (77 %).
L’évolution du nombre et du pourcentage par année d’incidents et d’effets indésirables en fonction de la gravité entre les années 2009 et 2013 est présentée dans le tableau AMPV2.
Tableau AMPV2. Évolution du nombre et du pourcentage d'incidents et d'effets indésirables en fonction de la gravité entre 2009 et 2013
|
Année de déclaration |
||||||
Type d'événement |
Gravité |
2009 |
2010 |
2011 |
2012 |
2013 |
2009 à 2013 |
Incidents |
Non graves |
43 (20,2%) |
53 (15,2%) |
47 (11,5%) |
52 (10,9%) |
82 (17,5%) |
277 (14,4%) |
Graves |
29 (13,6%) |
37 (10,6%) |
29 (7,1%) |
36 (7,5%) |
25 (5,3%) |
156 (8,1%) |
|
Effets indésirables |
Non graves |
4 (1,9%) |
22 (6,3%) |
20 (4,9%) |
23 (4,8%) |
31 (6,6%) |
100 (5,2%) |
Graves |
137 (64,3%) |
237 (67,9%) |
313 (76,5%) |
366 (76,7%) |
331 (70,6%) |
1384 (72,2%) |
|
Total |
213 (100%) |
349 (100%) |
409 (100%) |
477 (100%) |
469 (100%) |
1917 (100%) |
|
On observe, entre 2009 et 2013, une diminution des déclarations d’incidents graves et une augmentation des déclarations d’effets indésirables non graves. Cette augmentation est probablement liée au nombre croissant de déclarations d’hyperstimulations ovariennes (HSO) sans hospitalisation (considérées comme non graves) et attendues dans ce type de prise en charge. Les effets indésirables graves (gravité G3 et au-delà) représentant la majorité des événements indésirables rapportés.
Distribution des incidents et des effets indésirables en fonction de la typologie et de la gravité
La distribution des incidents et des effets indésirables selon la typologie déclarés en 2013 est présentée à la figure AMPV6.
Figure AMPV6. Distribution des incidents et effets indésirables déclarés en 2013 (n = 469)
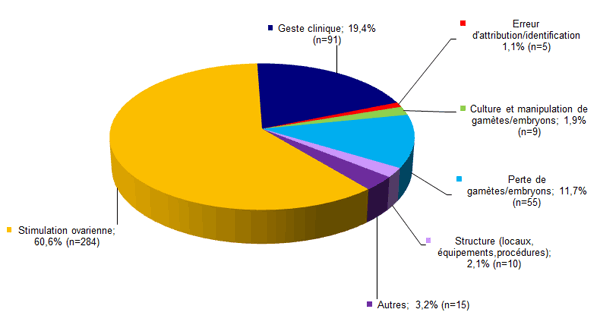
Pour l’année 2013, 284 événements indésirables (61 %) concernent la stimulation ovarienne, 91 (19 %) concernent un geste clinique lors de l’AMP (inséminations, ponctions, transferts), 55 (12 %) concernent la perte de gamètes/embryons, 10 (2 %) sont relatifs à la structure, 5 (1 %) concernent l’attribution des gamètes ou des embryons, 9 (2 %) concernent la culture des gamètes/embryons, et 15 (3 %) sont classés comme « autres ».
Incidents et effets indésirables relatifs à la stimulation ovarienne
La distribution des incidents et des effets indésirables relatifs à la stimulation ovarienne, en fonction de la gravité est présentée à la figure AMPV7.
Figure AMPV7. Nombre d’incidents et d’effets indésirables relatifs à la stimulation ovarienne en fonction de la gravité (2013, n = 284)
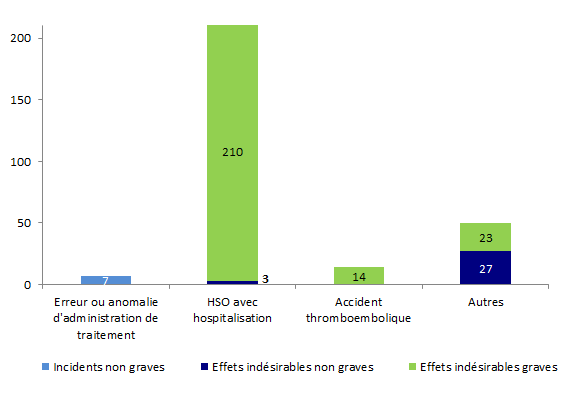
Plus de 60 % des événements indésirables rapportés concernent des événements relatifs à la stimulation ovarienne (n = 284). Ces 284 événements indésirables comprennent essentiellement des effets indésirables graves (87 %).
Il s’agit le plus souvent de syndromes d’hyperstimulation ovarienne sévères avec hospitalisation d’au moins 24 heures (203/284 soit 71 %).
Quatorze cas de thromboses ont été rapportés par 13 centres d’AMP chez des femmes âgées en moyenne de 36 ± 4,5 ans [28-42]. Il s’agit de 5 embolies pulmonaires dont 1 embolie pulmonaire bilatérale, 2 phlébites des membres inférieurs, 2 thromboses veineuses jugulaires internes, 1 thrombose de la veine sous-clavière, 1 thrombophlébite cérébrale, 1 papillophlébite de l’œil droit, 1 accident vasculaire cérébral ischémique et 1 hémiplégie gauche. Dans 5 observations, il est mentionné un syndrome d’hyperstimulation ovarienne sévère associé. Dans une observation, la thrombose est survenue à 10 semaines d’aménorrhée (SA) dans un contexte de grossesse gémellaire.
Parmi les 50 évènements renseignés comme « Autres », on retrouve 30% de torsions d’annexes (15/50) et 50% d’HSO sans hospitalisation (25/50).
Incidents et effets indésirables relatifs à un geste clinique lors de l’AMP
La distribution des incidents et des effets indésirables relatifs à un geste clinique lors de l’AMP est présentée à la figure AMPV8.
Figure AMPV8. Nombre d’incidents et d’effets indésirables relatifs à un geste clinique en fonction de la gravité (2013, n = 91)
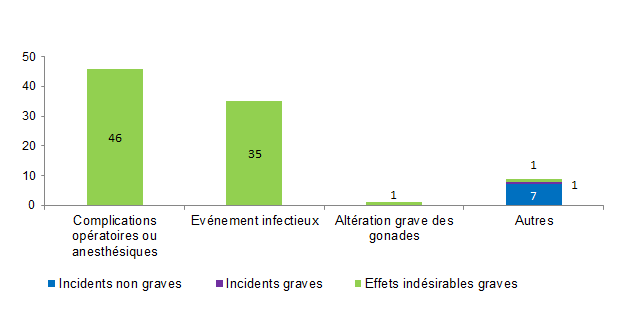
En 2013, 91 (19 %) événements indésirables sont en lien avec un geste clinique lors de l’AMP (insémination, ponction folliculaire, transfert embryonnaire, ...). Ces événements sont répartis en 83 effets indésirables graves et 8 incidents (1 incident grave et 7 incidents non graves).
Parmi les 91 événements associés à un geste clinique lors de l’AMP, 51% (46/91) concernent des complications opératoires ou anesthésiques tels que des hémopéritoines, des hématomes ovariens, des allergies médicamenteuses et 39 % (35/91) des infections.
Les événements renseignés comme « Autres » correspondaient par exemple, à un retard dans la prise en charge de la ponction entrainant l’annulation de la ponction, à une discordance entre le nombre d’ovocytes ponctionnés par rapport à celui attendu, ou à une erreur du nombre d’embryons à transférer.
Incidents relatifs à une perte ou destruction accidentelle partielle ou totale de gamètes / embryons
La distribution des incidents relatifs à une perte ou à une destruction de gamètes/embryons est présentée à la figure AMPV9.
Figure AMPV9. Nombre d’incidents relatifs à la perte ou à la destruction accidentelle de gamètes / embryons en fonction de la gravité (2013, n = 55)
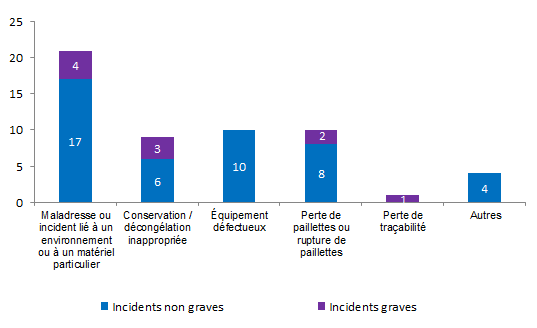
Cinquante-cinq incidents (12 %) rapportés en 2013 concernent des pertes ou destructions accidentelles partielles ou totales de gamètes ou d’embryons. Pour rappel, une perte totale des gamètes ou embryons sur la tentative est considéré comme un incident grave.
Il s’agit le plus souvent d’incidents non graves (82 %). Près d’une fois sur 2 ces incidents sont en rapport avec une maladresse ou avec un équipement ou un matériel défectueux.
Les événements renseignés comme « Autres » correspondaient par exemple, à la destruction par erreur d’embryons ou à la perte partielle d’ovocytes lors de la décoronisation.
Incidents relatifs à la structure
La distribution des incidents relatifs à la structure en fonction de la gravité est présentée à la figure AMPV10.
Figure AMPV10. Nombre d’incidents relatifs à la structure en fonction de la gravité (2013, n = 10)
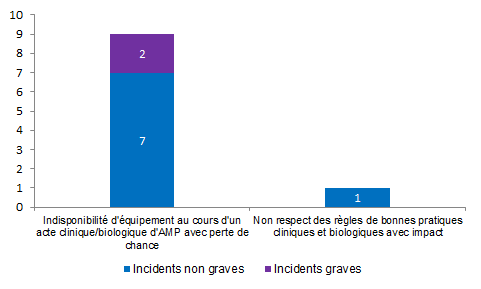
En 2013, 10 incidents relatifs à la structure ont été rapportés. Il s’agit uniquement d’incidents, le plus souvent non graves (80 %). La typologie la plus souvent rapportée concerne une indisponibilité d’équipement au cours d’un acte clinique/biologique d’AMP avec perte de chance.
Distribution des incidents et des effets indésirables en fonction de l’imputabilité
Selon la fiche de déclaration d’AMP vigilance et la méthodologie développée dans le guide de remplissage de la fiche de déclaration d’AMP vigilance, le déclarant doit évaluer l’imputabilité de l’incident ou de l’effet indésirable selon une échelle d’imputabilité proposée par l’Agence de la biomédecine.
La distribution des incidents et des effets indésirables en fonction de l’imputabilité est présentée à la figure AMPV11.
Figure AMPV11. Nombre d’incidents et d’effets indésirables en fonction de l’imputabilité (2013, n = 469)
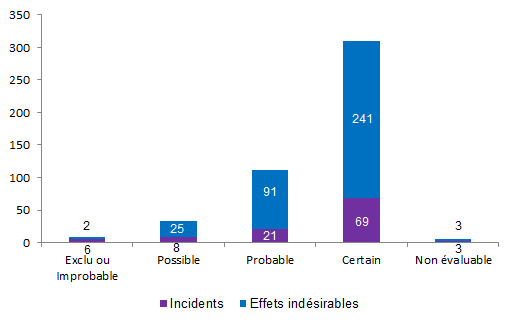
L’imputabilité a été cotée le plus souvent comme certaine (66%) à la fois pour les incidents (64%) et les effets indésirables (67%) en raison d’un lien fréquent entre l’activité et la survenue de l’évènement.
L’évaluation de l’imputabilité étant très variable d’un centre à un autre, cette information est donc difficile à interpréter. La définition de l’imputabilité doit être clarifiée.
Conséquences si l’événement indésirable concerne les gamètes, embryons et / ou tissus germinaux
En 2013 :
- parmi les 28 incidents déclarés concernant les gamètes, 17 ont entraîné une perte de gamètes ;
- parmi les 57 incidents déclarés concernant les embryons, 37 ont entraîné une perte d’embryons ;
- parmi les 3 incidents déclarés concernant les gamètes et les embryons, 3 ont entraîné une perte de gamètes et d’embryons ;
- aucun incident déclaré ne concerne les tissus germinaux.
Ces 57 incidents avec perte de gamètes et/ou d’embryons ont eu pour conséquences :
- une perte de chance de procréation totale sur la tentative pour 70 couples ou patients
- une perte de chance de procréation partielle sur la tentative pour 58 couples ou patients
- une perte de chance de procréation potentielle pour 86 couples ou patients.
Conséquences si l’événement indésirable concerne le patient (hospitalisation, mise en jeu du pronostic vital, invalidité/incapacité, effet délétère sur la fertilité, décès)
Le type de conséquences pour les 362 effets indésirables observés chez le patient est présenté à la figure AMPV12.
Le total des conséquences (390) est supérieur au nombre d’effets indésirables (362) dans la mesure où plusieurs conséquences sont parfois renseignées pour un même effet indésirable.Figure AMPV12. Type de conséquences pour les 362 effets indésirables observés chez le patient (2013)
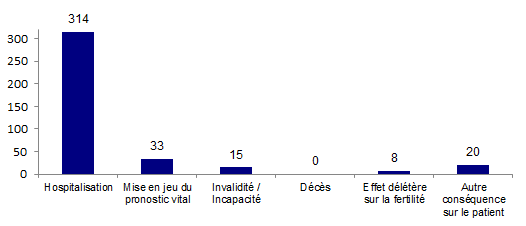
Pour 98% de ces conséquences (381/390), il s’agissait d’effets indésirables graves et pour 2% (9/390), il s’agissait d’effets indésirables non graves ayant eu des conséquences de type annulation du transfert ou congélation des embryons.
Parmi les 362 déclarations d’effets indésirables, la plupart (314/362 soit 87 %) ont entraîné une hospitalisation.
Les durées d’hospitalisation en fonction de la typologie de certains effets indésirables tels que les syndromes d’hyperstimulation ovarienne sévères, les thromboses, les complications opératoires ou anesthésiques, et les événements infectieux sont présentées dans le tableau AMPV3.Tableau AMPV3. Durée d'hospitalisation (jours) par type d'effet indésirable - 2013
Durée d'hospitalisation (jours) |
|||||||
Typologie des effets indésirables |
N* |
Moyenne |
Écart-type |
Médiane |
Min |
Max |
|
Événements relatifs à une stimulation ovarienne |
HSO avec hospitalisation |
203 |
6,3 |
4,2 |
5 |
1 |
28 |
Accident thromboembolique |
10 |
8,6 |
8 |
7 |
1 |
30 |
|
Autres |
20 |
4,4 |
7,4 |
2,5 |
1 |
35 |
|
Événements relatifs à un geste clinique |
Complications opératoires ou anesthésiques |
35 |
3,6 |
2,1 |
3 |
1 |
10 |
Evènement infectieux |
30 |
7,3 |
3,8 |
6,5 |
2 |
17 |
|
Altération grave des gonades |
1 |
13 |
NC |
13 |
13 |
13 |
|
Autres |
1 |
2 |
NC |
2 |
2 |
2 |
|
Total |
|
300 |
6 |
4,6 |
5 |
1 |
35 |
* : nombre d'effets indésirables pour lesquels la durée d’hospitalisation a été renseignée
NC : non calculable
Parmi les 314 déclarations d’effets indésirables ayant entraîné une hospitalisation, la durée d’hospitalisation a été renseignée pour 300 effets. Pour ces effets, la durée moyenne d’hospitalisation a été de 6 jours et une médiane de 5 jours. Parmi les 203 HSO avec hospitalisation, 8 HSO ont entrainé une hospitalisation d'une journée et 195 une hospitalisation de plus d'une journée.
A noter, la durée d’hospitalisation la plus longue a été de 35 jours chez une patiente de 34 ans hospitalisée pour aphasie et paresthésies de l’hémicorps droit en cours de traitement de stimulation en vue d’une FIV. Un diagnostic d’encéphalomyélite aigüe disséminée (ADEM) a été posé.
De façon cumulée, ces effets indésirables rapportés et pour lesquels on dispose de l’information, ont généré un nombre total de 1811 journées d’hospitalisation.
Actions mises en œuvre en 2013 suite à la survenue d'un événement indésirable (Partie A de la fiche de déclaration)
La distribution du nombre de déclarations par type d’actions mises en œuvre est présentée dans la figure AMPV13.
Figure AMPV13. Actions mises en œuvre en 2013 suite à la survenue d’un évènement indésirable
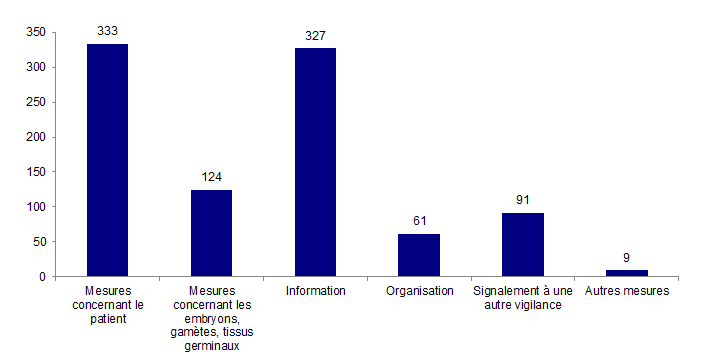
Dans 85 % des déclarations (398/469), au moins une action de prise en charge thérapeutique ou préventive a été prise. Il s’agit dans 84% (333/398) de mesures concernant les patients et dans 82 % (327/398) de mesures concernant l’information.
Mesures concernant le patient
Parmi les 333 déclarations ayant comporté des mesures concernant le patient, le plus souvent il s’agit de la prise en charge comprenant un traitement spécifique et/ou des examens complémentaires et/ou une surveillance et/ou une prise en charge psychologique (n=292).
Mesures concernant les embryons, gamètes, tissus germinaux
Parmi les 124 déclarations ayant comporté des mesures concernant les embryons, gamètes et tissus germinaux, le plus souvent l’action prise concernait la congélation des embryons, gamètes ou tissus germinaux (n=84).
Mesures concernant l’information
Pour 327 déclarations, il y a eu une information spécifique par rapport à l’événement indésirable. L’information a été communiquée le plus souvent au patient (n = 298) ou au personnel (n = 180), plus rarement à la direction de l’établissement (n = 49) ou à l’ARS (n = 3).
Mesures concernant l’organisation
Parmi les 61 déclarations ayant comporté des mesures concernant l’organisation, la survenue de l’événement a entrainé le plus souvent la mise en place ou la modification de procédures (n=44).
Mesures concernant le signalement à une autre vigilance
Parmi les 469 déclarations, le centre d’AMP a signalé à une autre vigilance dans 91 cas dont 61 fois à la pharmacovigilance, 12 fois à la matériovigilance, 5 fois à la biovigilance et 22 à un autre système de vigilance ou de surveillance sanitaire (coordination / pôle des vigilances de l’hôpital, gynerisk, identito-vigilance, service qualité de l’établissement, comite morbimortalité).
Ces transmissions à d’autres vigilances représentent 19% des déclarations, ce qui reflète la dimension transversale importante de l’AMP vigilance et nécessite donc une coordination avec les autres systèmes de vigilance que ce soit au niveau de l’établissement ou au niveau national.
De plus, l’Agence de la biomédecine a transmis 5 déclarations à une autre vigilance dont 4 en matériovigilance et 1 en pharmacovigilance.
Évitabilité et Maitrise des événements indésirables
Pour 23% des déclarations (110/469), le centre d’AMP a estimé que l’événement indésirable était évitable. Il faut noter que pour la moitié des déclarations (51 %), cet item n’a pas été renseigné ou a été renseigné « Ne sait pas ».
Parmi les déclarations pour lesquelles l’item « événement maitrisé » (Partie B de la fiche de déclaration) a été renseigné, on observe, entre 2009 et 2013, une augmentation du taux d’événements maitrisés passant de 89,6 % en 2009 à 97,1 % en 2013.
Pour plus d’informations, le rapport annuel d’AMP vigilance 2013 adressé au ministre en charge de la santé est téléchargeable à partir de juillet 2014 sur le site de l’Agence de la biomédecine (http://www.agence-biomedecine.fr/professionnels/amp-vigilance.html).
1 Décret n°2008-588 du 19 juin 2008 transposant en matière de don de gamètes et d’assistance médicale à la procréation la directive 2004/23/CE du Parlement européen et du Conseil du 31 mars 2004
2 Ces laboratoires sont chargés de préparer le sperme en vue de l’insémination artificielle
3 La date de déclaration correspond à l’envoi de la Partie A de la fiche de déclaration (Signalement immédiat)
4 Le nombre d'actes d'AMP comprend le nombre de tentatives (cycles d'insémination artificielle (IIU, IIC) ; ponctions d'ovocytes dans le cadre des fécondations in vitro (FIV, ICSI) ; transferts d'embryons congelés (TEC)), le nombre de patients ayant fait une nouvelle autoconservation dans l’année (en cours d’AMP et pour la préservation de la fertilité) et le nombre de donneuses d’ovocytes et de donneurs de spermatozoïdes.